 St John
St John 
One of the most exciting applications of advancements in artificial intelligence has been the modelling of protein structures and protein folding. Last year DeepMind announced their AlphaFold project, which claims to solve a 50-year-old grand challenge in biology - the protein folding problem. Christian Anfinsen famously posited that a protein sequence should fully determine the structure of the protein. In other words, given a sequence of amino-acids, one should be able to predict how that protein will ‘fold’ to form a 3D structure associated with its function. This is an incredibly interesting and rich area of research, spanning multiple domains and requiring immense computing power. With that said, this is not exactly what I’m writing about here. The application I’m interested in is applying the immense power of pre-trained, unsupervised language models - like ProtBert - to ‘skip’ the protein folding ‘step,’ and go directly from a protein sequence to its predicted protein function.
At a surface level this seems like an inplausible - perhaps impossible - task. Anyone familiar with recent successes of language models will be unsurprised to hear that natural language processing methods have had great success in applications involving protein sequences. One can think of a protein sequence as a recipe which defines rules about how a protein should ‘behave’ and carry out some function(s) in the language of amino acids. This is exactly what we will exploit by using models that are ‘aware’ of the context in which a particular amino acid is placed. This is crucial in the protein folding problem, as how a protein folds is highly dependent on the context in which the amino acids in its sequence are found.
Let’s jump striaght into it. To get the highest performance out of language models for the protein function prediction problem, we will be using open-source models that have been partially trained in the context of protein sequences. This is where DeepChain and associated open-source repos come in.
DeepChain
DeepChain is InstaDeep’s AI protein design platform which allows users to make use of incredibly powerful, open-source language models to design and interact with proteins. Of interest to us is their open-source bio-datasets and bio-transformers packages which allow us to use state-of-the-art deep learning models for almost any task imaginable. People with coding experience can develop their own applications for the DeepChain platform by deploying apps to the DeepChain Hub. The template defining how such an app should be laid out is available on GitHub. We will make use of exactly this platform for quickly developing a state-of-the-art protein function prediction model which leverages open-source, pretrained language models.
Let’s set up our development environment for DeepChain. I highly recommend making use of Anaconda for ensuring compatible dependencies and avoiding
frustrating issues with other coding projects. We can start off by creating a new Anaconda environment with conda create --name deepchain-env python=3.7 -y.
With this done, we can activate the environment with conda activate deepchain-env, and then download the necessary packages for running DeepChain apps using pip install deepchain-apps.
This might take a while to set up, so go grab yourself a cup of tea. Thankfully, you should only have to do this once! The next step is using the deepchain CLI to connect with
and interface with the DeepChain platform. To login to DeepChain from command line we will need a personal access token (PAT) which you can find in your profile section of the DeepChain
website. Once you’ve copied this token, simply use deepchain login and paste your PAT. From here we are cruising. Just enter deepchain create YourApplicationName and we are
ready to start developing.
Gene Ontology Prediction
The task I am interested in is taking a protein sequence and mapping it to associated protein function. This is a notoriously difficult task since proteins can code for multiple tasks, and different proteins can code for similar functions. This is a dramatic oversimplification, but it captures our task fairly well. Ultimately, we are going to try predict The Gene Ontology given only a protein sequence.
My plan was to work with a unsupervised prediction method presented by A. Villegas-Morcillo et. al in a paper called Unsupervised protein embeddings outperform hand-crafted sequence and structure features at predicting molecular function. Coming up with plausible - yet fairly simple - ideas can be tricky. I started by just doing web searches for deep learning methods for function prediction and stumbled across this paper. To ensure I was working with reasonable research, I mapped out the research knowledge graph using Connected Papers. This way I got a picture of how this work relates to major works in the field. I briefly read some details in major related papers to see what the common approaches to this problem were. During this research, I came across the BLAST algorithm, which is a more ‘brute force’ approach to the task of finding similarities between proteins and, by extension, function.
All of this would be unsuprising to anyone familiar with the field - to say the least. I should stress the point of this project was to display how someone with some basic technical knowledge can get started developing very competitive applciations in technical fields they have no previous domain knowledge in. With that said, let’s consider the data used in the paper.
The Dataset & Embeddings
The dataset used during development of this app is largely based on the subset of Protein Data Bank (PDB) data provided by the paper. The authors lay out the details of how they filtered appropriate sequences for use with their models. The main points to consider are that they “considered proteins with sequence length in the range [40, 1000] that had GO annotations in the Molecular Function Ontology (MFO) with evidence codes ‘EXP’, ‘IDA’, ‘IPI’, ‘IMP’, ‘IGI’, ‘IEP’, ‘HTP’, ‘HDA’, ‘HMP’, ‘HGI’, ‘HEP’, ‘IBA’, ‘IBD’, ‘IKR’, ‘IRD’, ‘IC’ and ‘TAS.’”
The original paper also considers other subsets of common datasets, like SwissProt. This could be an interesting area for expansion of this app. Training on a larger dataset could be very useful in this case. We should note that different datsets have different associated Gene Ontology labels associated with the proteins. This is important as it defines the layer sizes we want in our simple MLP scenario. In the PDB case, we have 256 possible classes a protein can be assigned to.
Another crucial design decision is in how to take a raw sequence and map it to some embedding. What we are basically doing here is trying to capture the ‘essence’ of the protein sequence by considering the order and context of the emino acids in the sequence. Based on what sequences the model has seen before, it will map the sequence to some latent representation. This representation of the sequence is much simpler for a simple model to train on as we can fix the size of the embedding. In this case I make use of ProtBert to output a [1,1024] dimensional embedding for each protein. This means that we can take any arbitrary length sequence and map it to an appropriate input size for our model. The ProtBert model was pretrained on Uniref100, a dataset consisting of 217 million protein sequences.
The Application and Model
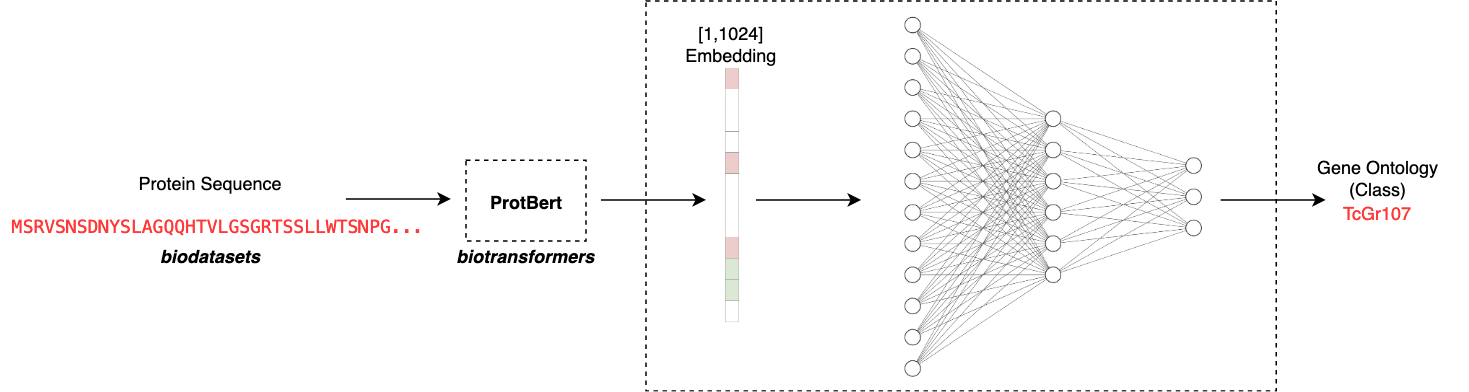
The app is designed to be very easy to use. All one needs to do is input a chosen amino acid sequence and the app will automate computing an embedding using the biodatasets ProtBert model. This can be run as follows:
1
2
3
4
5
6
7
8
9
10
11
seq = [
"PKIVILPHQDLCPDGAVLEANSGETILDAALRNGIEIEHACEKSCACTTCHCIVREGF \
DSLPESSEQEDDMLDKAWGLEPESRLSCQARVTDEDLVVEIPRYTINHARE",
"PMILGYWNVRGLTHPIRLLLEYTDSSYEEKRYAMGDAPDYDRSQWLNEKFKLGLDFPN \
LPYLIDGSRKITQSNAIMRYLARKHHLCGETEEERIRVDVLENQAMDTRLQLAMVCYS \
PDFERKKPEYLEGLPEKMKLYSEFLGKQPWFAGNKITYVDFLVYDVLDQHRIFEPKCL \
DAFPNLKDFVARFEGLKKISDYMKSGRFLSKPIFAKMAFWNPK"
]
app = App(device = "cpu") # Set to App() for GPU training
score_dict = app.compute_scores(seq)
print(score_dict)
The app then outputs a numpy array with scores corresponding the the classes - and thus function - it believes the protein sequence codes for. Since protein sequences can code for multiple functions, and multiple proteins can code for similar functions, there will likely be a range of scores! An example of output for the above sequences is:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
{
'Class':
tensor(
[[ 0.0256, -0.0338, -0.0292, -0.0446, -0.0345, 0.0050, 0.0508, -0.0102,
... 0.0110, 0.0077, 0.0633, 0.0896, -0.0260, 0.0021, -0.1576, -0.0036],
[ 0.1083, -0.1110, -0.0437, -0.0807, 0.0331, -0.0005, 0.0704, -0.0314,
... -0.0331, 0.0576, -0.0238, 0.0333, -0.0444, -0.0256, -0.1072, -0.0553]],
grad_fn=<AddmmBackward>),
'Embedding':
tensor(
[[0.0000, 0.0000, 0.0000, ..., 0.0000, 0.1248, 0.0276],
[0.0000, 0.1116, 0.0000, ..., 0.0000, 0.0000, 0.0356]],
grad_fn=<MulBackward0>)
}
The Metrics
Of course, if you want to use - or, especially, modify - this app, it is crucial to understand what exactly the app is telling you. The default metric I implemented was the maximum protein-centric F-measure/F-score (\(F_{max}\)). This basically tells us about the trade-off between the precision and recall of our model. For the less statistics-inclined, this informs us of two things; (1) how many of the predicted GO classes are relevant in reality, and (2) how many of the relevant classes did our model capture? Even simpler, what did our model incorrectly predict and what did our model incorrectly miss? In this sense, it is a simple way to capture a total measure for thinking about false/true negatives/positives.
The authors of the paper include two more measures. These include the normalized minimum semantic distance (\(S_{min}\)) and the term-centric ROC-AUC. If these are not familiar to you, I highly recommend reading up on these. They are extremely useful for classification tasks in general. If you’re thinking about some ways to extend this app, take a look at the code I’ve included for these measures (from the original paper) and try implement these.
Future Work
Some other ideas I had were using a CRISPR dataset for cancer genes that had been screened. This led me to the Cancer Dependency Map project being run by the Sanger Institute. This project aims to identify dependencies between all cancer cells. My thinking was that we could take in a gene or protein sequence and predict relation to known cancer dependencies. Perhaps this could help researchers design cancer therapeutics using InstaDeep’s DeepChain platform.
This app has loads of room for improvement. For one, note that the original paper made use of ELMo embeddings for training and evaluating their models. They also made use of a wide variety of different models, and used fairly (empirically) rigorous procedures for testing their models. For example, they performed 5-fold cross validation. They also were able to train on the full SwissProt dataset. This dataset can be added in the future, but calculating the embeddings on a local machine will take a fair amount of compute. Feel free to do this and add the embeddings to the open access biodatasets portal 😄 🧬.
This app is open to open source contributions. Please connect with me on the public GitHub repo to discuss ideas for making this app more generally useful. Some ideas for expansion involve providing more models for training the multi-label classification task. The original paper using unsupervised embeddings for GO classification compared multiple models, including the MLP presented here. Some other interesting models include GNNs and models that combine sequence information with 3D structural information.
